Causas genéticas de diabetes de início precoce: do diabetes neonatal às lipodistrofias
Assista ao vídeo e leia o conteúdo abaixo.
Diabetes monogênico
Define-se o diabetes mellitus (DM) como um grupo de doenças metabólicas genética e genotipicamente heterogêneas, de ampla prevalência mundial, caracterizadas por hiperglicemia decorrente de defeitos na secreção ou na ação da insulina. As formas mais comuns, o DM tipo 1 e o DM tipo 2, têm etiologias associadas a uma variedade de diferentes genes, mas nenhum isoladamente é suficiente para causar o quadro. Entretanto, estima-se que aproximadamente 1% dos pacientes com diabetes tenha uma das formas monogênicas da doença, derivadas de variantes alélicas patogênicas em um único gene.
Sua forma mais comum é chamada de MODY, sigla, do inglês, de maturity onset diabetes of the young, cujos subtipos MODY-GCK (MODY 2) e MODY-HNF1A (MODY 3) respondem por quase 70% de tais casos.
O desafio clínico é identificar esses casos e, especialmente, determinar quem pode se beneficiar dos estudos genéticos e, ainda, quais genes devem ser analisados (veja quadro).
Classificação do MODY em subtipos de acordo com a alteração genética envolvida
| Tipo | Gene envolvido | Frequência entre os casos de Mody | Características fenotípicas associadas |
| MODY 1 | HNF4A | 2-10% | - Macrossomia ao nascimento - Hipoglicemia neonatal transitória - Hiperglicemia progressiva - Idade de início: adolescência/adulto jovem - HDL baixo - Sensibilidade a sulfonilureias |
| MODY 2 | GCK | 20-63% | - Hiperglicemia leve, assintomática e estável - Idade de início: ao nascimento/infância - Pequeno incremento no teste de tolerância oral à glicose de duas horas - Complicações microvasculares raras - Tipicamente não necessita de tratamento |
| MODY 3 | HNF1A | 21-72% | - Hiperglicemia progressiva - Idade de início: adolescência/adulto jovem - Baixo limiar renal de reabsorção de glicose - Grande incremento no teste de tolerância oral à glicose de duas horas - HDL alto - Sensibilidade a sulfonilureias |
| MODY 4 | PDX1 | <1% | - Diabetes e agenesia parcial do pâncreas |
| MODY 5 | HNF1B | 1-6% | - Hiperglicemia progressiva - Disfunção renal/cistos renais - Hiperuricemia/hipomagnesemia - Malformações urogenitais - Disfunção pancreática exócrina subclínica - Transaminases elevadas |
| MODY 6 | NEUROD1 | <1% | - Poucos casos descritos |
| MODY 7 | KLF11 | Raro | - Poucos casos descritos |
| MODY 8 | CEL | Raro | - Poucos casos descritos - Associado à atrofia pancreática e à dislipidemia - Disfunção pancreática exócrina |
| MODY 9 | PAX4 | Raro | - Poucos casos descritos |
| MODY 10 | INS | Raro | - Poucos casos descritos |
| MODY 11 | BLK | Raro | - Poucos casos descritos |
| MODY 12 | ABCC8 | Raro | - Sensibilidade a sulfonilureias |
| MODY 13 | KCNJ11 | Raro | - Sensibilidade a sulfonilureias |
| MODY 14 | APLL1 | Raro | - Poucos casos descritos |
Adaptado de: Genetic Diagnosis of Endocrine Disorders, 2016; 21-30.
Diabetes mellitus neonatal
Caracterizado pelo diagnóstico precoce da condição, antes dos 6 meses de vida, o diabetes mellitus neonatal (DMN), também conhecido por diabetes neonatal, tem prevalência estimada em 1:90.000 a 1:160.000 nascidos vivos. Pode ser permanente ou transitório, quando entra em remissão nos primeiros anos da infância, embora com possibilidade de recorrer mais tardiamente. Em 80% a 85% dos casos, a forma monogênica responde pelo quadro. É importante considerar que algumas crianças com diabetes após 6 meses de idade também terão a doença monogênica, especialmente quando os autoanticorpos pancreáticos forem negativos.
O DMN pode ser resultante do prejuízo no desenvolvimento ou na função da célula betapancreática ou de sua destruição progressiva e manifesta-se de modo isolado ou como parte de síndromes multissistêmicas, quando se associa à epilepsia, ao atraso do desenvolvimento neuropsicomotor e ao hipotiroidismo, entre outras. A herança genética pode ser autossômica ou ligada ao X, dominante ou recessiva.
Todas as crianças diagnosticadas com diabetes nos primeiros seis meses de vida devem ser avaliadas com testes genéticos para diabetes neonatal. A confirmação genética é possível em aproximadamente 80% dos casos, até porque mais de 25 diferentes genes já foram relacionados a essa condição.
O diagnóstico molecular precoce é clinicamente relevante para determinar o tipo de tratamento, pois algumas formas de diabetes neonatal respondem às sulfonilureias.
Cerca de metade das crianças com DMN evolui com um quadro permanente, enquanto a outra metade apresenta remissão semanas a meses após o início do quadro clínico, mas com elevadas chances de recorrência (veja tabela abaixo, de evolução e prevalência do DMN).
Subtipos de diabetes neonatal e principais genes envolvidos
- Anormalidades do desenvolvimento do pâncreas: PLAGL1, ZFP57 (síndrome da hipometilação múltipla), PDX1, HNF1B, GATA6, NEUROD1
- Anormalidade na função da célula betapancreática: KCNJ11, ABCC8, GCK
- Destruição da célula betapancreática: INS, FOXP3 (síndrome autoimune), WFS1 (síndrome de Wolfran), EIF2AK3
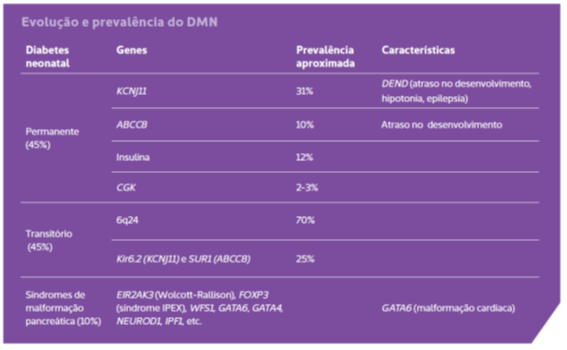
Clique na tabela para ampliar
Lipodistrofias
As síndromes de lipodistrofia são condições heterogêneas herdadas ou adquiridas, extremamente raras, caracterizadas por perda seletiva de tecido adiposo subcutâneo e armazenamento lipídico ectópico. O acúmulo de gordura ocorre especialmente no fígado e no músculo, causando resistência insulínica e suas complicações – diabetes mellitus, hipertrigliceridemia, acantose nigricante, hipertensão, síndrome dos ovários policísticos e doença hepática gordurosa não alcoólica.
Essas entidades são categorizadas de acordo com a etiologia (genética ou adquirida) e com a extensão da perda de tecido adiposo, que pode afetar o corpo inteiro (generalizada) ou apenas algumas regiões (parcial).
A síndrome de lipodistrofia engloba quatro categorias principais: generalizada congênita, parcial familiar, generalizada adquirida e parcial adquirida. Subtipos adicionais incluem distúrbios progeroides e autoimunes, entre outros, como em pacientes infectados pelo HIV ou a forma localizada, ocasionada por drogas injetáveis. Os dois subtipos mais prevalentes de lipodistrofia genética são a congênita generalizada e a parcial familiar, cada qual com relatos de 300 a 500 pacientes no mundo todo.
As principais causas de mortalidade incluem a doença cardíaca (cardiopatia, insuficiência cardíaca, infarto do miocárdio, arritmia), a doença hepática (insuficiência hepática, hemorragia gastrointestinal, carcinoma hepatocelular), a insuficiência renal, a pancreatite aguda e a sepse.
Classificação das lipodistrofias genéticas
Autossômica recessiva (AR)
- Lipodistrofia generalizada congênita (síndrome de Berardinelli-Seip): AGPAT2, BSCL2, CAV1, PTRF
- Displasia mandibuloacral: LMNA, ZMPSTE24, SPRTN, WRN, BANF1
- Lipodistrofia parcial familiar: CIDEC, LIPE, WRN, PCYT1A, PPARG
- Lipodistrofia autoinflamatória: (síndrome JMP/CANDLE): PSMB8
- Fenótipos LGC-like: PPARG, FOS
Autossômica dominante (AD)
- Lipodistrofia parcial familiar: LMNA, PPARG, AKT2, PLIN1
- Progéria ou síndrome de Hutchinson-Gilford: LMNA
- Síndrome progeroide atípica: LMNA
- Síndrome progeroide neonatal: FBN1, CAV1
- Hipoplasia mandibular, surdez e características progeroides: POLD1
- Lipodistrofia associada à síndrome Short, do inglês, short stature, hyperextensibility, hernia, ocular depression, rieger anomaly and teething delay: PIK3R1
- Lipodistrofia associada à síndrome de Keppen-Lubinsky: KCNJ6
Características clínicas que aumentam a suspeição de lipodistrofia
- Característica essencial: Ausência generalizada ou regional de gordura corporal
- Características físicas:
- - Atraso no desenvolvimento
- Músculos proeminentes
- Flebomegalia
- Acantose nigricante
- Xantomas
- Aparência acromegaloide ou progeroide - Comorbidades associadas:
- - Diabetes mellitus com necessidade de altas doses de insulina (≥2 U/kg/dia)
- Hipertrigliceridemia grave
- História da pancreatite aguda secundária à hipertrigliceridemia
- Esteato-hepatite não alcoólica em indivíduo não obeso
- Miocardiopatia de início precoce
- Síndrome dos ovários policísticos - Padrão de herança AD ou AR das características físicas ou complicações metabólicas
- Hiperfagia significativa
Diagnóstico
Convém levantar a suspeita de lipodistrofia em pacientes com perda progressiva de tecido adiposo subcutâneo, observando-se sua distribuição, bem como a presença de alguns sinais, como músculos proeminentes, flebomegalia, acantose nigricante, hepatomegalia, xantomas e aparência acromegaloide ou progeroide. A antropometria convencional e a densitometria de corpo inteiro podem ser realizadas para confirmar o padrão de perda de gordura.
Quanto aos exames laboratoriais, estão indicadas as dosagens do complemento e dos autoanticorpos para o diagnóstico das síndromes de lipodistrofia adquirida. Como os testes de leptina no soro não são padronizados e as concentrações dessa substância em pacientes com lipodistrofia, especialmente nas formas parciais, se sobrepõem às da população geral, essa mensuração não contribui com o diagnóstico, mas pode direcionar a escolha do tratamento. Já o teste genético tipo MODY expandido pode confirmar os casos suspeitos de lipodistrofia familiar.
O diagnóstico diferencial deve incluir condições que apresentem grave perda de peso, como desnutrição, anorexia nervosa, diabetes mellitus não controlado, tirotoxicose, insuficiência adrenocortical, caquexia do câncer, desnutrição associada ao HIV e infecções crônicas.
Conheça os painéis multigênicos que o Fleury realiza para o diagnóstico de diabetes monogênico
 |  |
Clique nas tabelas para ampliar
Consultoria médica
Dr. José Viana Lima Junior
Dra. Maria Izabel Chiamolera
Dra. Milena G. Teles Bezerra
Dr. Pedro Saddi
Dra. Rosa Paula M. Biscolla
Outros artigos
Investigação por imagem da dor abdominal recorrente em adolescente
Ultrassonografia auxilia o diagnóstico, métodos confirmam a suspeita e definem a extensão do quadro
Por que precisamos de vacinas anuais contra a gripe e a Covid-19?
A revacinação é a solução para conter o crescente número de casos dessas infecções.
Fleury apresenta seu novo Centro Integrado de Neurologia
Toda a inovação tecnológica em diagnóstico neurológico num só ambiente
Infertilidade primária, com falhas de implantação embrionária
Que hipótese cogitar diante das imagens histeroscópicas?