A evolução do diagnóstico dos erros inatos do metabolismo
No início do século passado, o médico inglês Sir Archibald Garrod atendeu um bebê que apresentava urina escura. Investigando o quadro em outras famílias, ele descobriu a via metabólica associada à alcaptonúria e sua relação com uma herança mendeliana. Já em 1934, uma mãe procurou o médico Asbjörn Fölling porque seus dois filhos apresentavam um odor estranho e deficiência intelectual, o que culminou na primeira descrição da fenilcetonúria. Duas décadas depois, Bickel e sua equipe introduziram, para esses pacientes, a restrição da fenilalanina na alimentação, que se mostrou um tratamento eficaz.
O conhecimento de que uma deficiência intelectual poderia ser prevenida por um diagnóstico precoce e pelo tratamento dietético levou ao desenvolvimento do primeiro programa de triagem neonatal, em 1960, pelo biólogo Robert Guthrie. A partir de então, o projeto se expandiu ao redor do mundo até um ponto em que, com o uso de tecnologias cada vez mais modernas, é possível triar precocemente um grande número de doenças raras (1:1.500 nascimentos), conhecidas como erros inatos do metabolismo (EIM).
De etiologia genética, os EIM associam-se à deficiência de componentes intracelulares importantes para diversas vias metabólicas, como enzimas, cofatores e transportadores, o que pode ocasionar o acúmulo de uma substância tóxica ou a redução da capacidade de síntese de compostos essenciais ou da produção de energia.
Essas condições exibem amplo espectro clínico e geralmente afetam múltiplos sistemas. Embora os sintomas possam se iniciar em qualquer idade – desde intraútero até a fase adulta –, cerca de 80% dos casos se manifestam nos dois primeiros anos de vida.
As alterações neurológicas, incluindo crises epilépticas, distúrbios do movimento, deficiência intelectual e hipotonia, entre outras, são sinais comuns encontrados nos pacientes. Tanto é assim que os EIM respondem por cerca de 5% a 10% dos casos de distúrbios do desenvolvimento intelectual de etiologia genética. Já outras manifestações sistêmicas ocorrem em cerca de 70% dos quadros, tendo as disfunções hepática e renal, os distúrbios do crescimento, as alterações hematológicas e as manifestações cardíacas, cutâneas e osteoarticulares como os principais sintomas.
Período pré-natal
Algumas anormalidades detectadas na ultrassonografia obstétrica podem indicar a possibilidade de um EIM ainda na gestação. A hidropsia fetal, por exemplo, pode ser identificada em fetos com doenças do depósito lisossômico, como as mucopolissacaridoses, a doença de Gaucher e a gangliosidose GM1. Para toda suspeita de doença genética no pré-natal, recomenda-se a confirmação por amniocentese ou biópsia de vilo corial.
Recém-nascidos
Muitos EIM se manifestam ainda no primeiro mês de vida. Contudo, a maioria dos neonatos acometidos aparenta-se saudável ao nascimento, visto que os metabólitos decorrentes do quadro podem ser depurados pela circulação placentária durante o período intrauterino. O acúmulo dessas substâncias começa apenas após o nascimento, com um período assintomático de duração variável.
Os sintomas nos recém-nascidos são inespecíficos e incluem letargia, sucção débil, vômitos, respiração anormal, hipotonia e crises epilépticas, só para citar alguns. Tais sinais se confundem com os de um quadro infeccioso nessa faixa etária. Assim, devem levantar a suspeita de uma doença metabólica especialmente se associados a condições como hipoglicemia inexplicada, alterações da função hepática, acidose metabólica e encefalopatia ou, então, a uma má resposta ao tratamento antibiótico. Nesses casos, a hipótese de EIM tem de ser aventada mesmo em um bebê com resultados normais na triagem neonatal.
Crianças
A maior parte das doenças apresentadas no período neonatal costuma se manifestar de forma similar ou com menor gravidade durante a infância. Nessa fase, podem ser notadas características dismórficas associadas ao quadro, que não são de fácil identificação ou que não estavam presentes ao nascimento. O atraso global do desenvolvimento ou a regressão de habilidades já adquiridas configuram sinais importantes nesse grupo etário.
Adolescentes e adultos
Ocasionalmente, um EIM pode se manifestar apenas na adolescência ou na fase adulta, como as doenças de depósito lisossômico de origem no adulto. Tais pacientes podem ter uma atividade enzimática residual que possibilita um acúmulo muito lento de metabólitos tóxicos, com sintomas mais tardios.
Diagnóstico desafiador
O diagnóstico dos EIM constitui um desafio na prática clínica devido ao número e à variedade das doenças desse grupo, que têm fenótipo amplo e inespecífico, mimetizando até mesmo condições mais comuns.
Assim, o primeiro passo para direcionar a solicitação de exames laboratoriais é determinar se a enfermidade está relacionada a defeitos no metabolismo de pequenas moléculas ou de organelas. Pacientes com doenças de pequenas moléculas costumam apresentar quadro agudo, precisando de intervenções de emergência. Já aqueles com alteração do metabolismo de organelas comumente cursam com manifestações neurológicas ou neuromusculares progressivas, organomegalias e disfunções hepáticas, associadas ou não a dismorfismos. Contudo, estas últimas também podem eventualmente levar a crises metabólicas, sendo a hipoglicemia e a acidose as descompensações mais frequentes.
Muitos exames laboratoriais têm utilidade como marcadores de estresse metabólico e, portanto, devem ser solicitados, no início, para todos os pacientes com suspeita de um possível EIM. Os resultados desses testes auxiliam o raciocínio clínico, a identificação de uma categoria de doenças e o direcionamento para a escolha de outros métodos diagnósticos mais específicos (tabela 1). Nesse sentido, as pesquisas de aminoácidos no plasma e de ácidos orgânicos na urina e o perfil de acilcarnitinas (tabela 2), assim como a pesquisa de ácidos graxos de cadeia muito longa e a dosagem de carnitina, configuram os testes mais úteis.
Em casos particulares, a medida de atividades enzimáticas, a análise histopatológica, a avaliação do líquido cefalorraquidiano e os estudos de Neuroimagem, com destaque para a ressonância magnética (RM) e para a RM com espectroscopia, podem ser necessários para alcançar o diagnóstico específico.
| Tabela 1. Exames úteis para investigação inicial na suspeita de um EIM | |
| Sangue | |
| Hemograma | Mostra alterações presentes em alguns EIM, sobretudo em defeitos no metabolismo de organelas. Uma pancitopenia, por exemplo, fala a favor de doença de Gaucher. |
| Função hepática, incluindo coagulograma | Sugere alterações presentes em diversos quadros, como defeitos de oxidação de ácidos graxos, tirosinemia tipo 1 e intolerância hereditária à frutose. |
| Função renal, eletrólitos e gasometria (equilíbrio ácido-básico) | A presença de um quadro de acidose metabólica com aumento do ânion gap pode estar relacionada com o acúmulo de ácidos orgânicos. O achado deve ser avaliado em conjunto com outros parâmetros metabólicos, como glicemia, cetose, lactatemia e amonemia. As acidemias orgânicas são o principal grupo de doenças que cursam com acidose metabólica e cetose, enquanto a presença de acidose e hipoglicemia sem cetose pode ser encontrada nos defeitos de oxidação dos ácidos graxos mitocondriais. |
| Glicose | Muitas doenças metabólicas se caracterizam por hipoglicemia grave, desde os quadros mais típicos que afetam o metabolismo dos carboidratos, como as doenças de depósito do glicogênio, os defeitos de gliconeogênese e os de oxidação mitocondrial de ácidos graxos, até as acidúrias orgânicas e algumas aminoacidopatias. Na avaliação desses casos, deve-se considerar se há cetose associada ao quadro. Ademais, obter a história do tempo decorrido entre a alimentação e a evolução para hipoglicemia pode auxiliar o raciocínio diagnóstico – uma hipoglicemia pós-prandial ou após curto período de jejum está geralmente relacionada à superutilização da glicose, como ocorre no hiperinsulinismo, enquanto a hipoglicemia após jejum prolongado (acima de 8h) sugere defeito de oxidação dos ácidos graxos. Uma queda dos níveis glicêmicos em um período intermediário (4-8h de jejum), por sua vez, é mais característica de uma glicogenose ou de uma doença que afeta a gliconeogênese. |
| Amônia | O nível plasmático de amônia deve ser dosado em todo paciente com alteração do nível de consciência ou encefalopatia, em especial crianças. A hiperamonemia pode ser causada por muitas condições não metabólicas, incluindo doença hepática, shunt porto-cava e intoxicação por ácido valproico, entre outras. Contudo, uma elevação importante, de 10 a 100 vezes o limite superior de normalidade, pode se associar a defeitos do ciclo da ureia. Outros EIM, como as acidemias orgânicas e os defeitos mitocondriais de oxidação dos ácidos graxos, também resultam em hiperamonemia, mas, em geral, menos significativa. |
| Lactato | O equilíbrio fisiológico do ácido lático circulante pode ser perturbado por condições metabólicas e não metabólicas. A acidose láctica é mais frequentemente provocada por hipóxia tecidual devido a um suprimento inadequado de oxigênio, que pode ocorrer por causas diversas, como choque hipovolêmico ou cardiogênico. Tais quadros não costumam ser acompanhados de cetose. Uma vez excluídas etiologias não metabólicas de hiperlactatemia, as causas mais comuns são os defeitos de oxidação dos ácidos graxos, as acidemias orgânicas e, mais raramente, os defeitos do ciclo da ureia. Outros quadros relacionados a uma elevação persistente de lactato são as doenças que afetam a gliconeogênese ou o armazenamento do glicogênio, o ciclo de Krebs ou o metabolismo do ácido pirúvico. |
| Creatinoquinase (CK total) | Associa-se à cardiomiopatia e pode estar elevada em condições metabólicas como defeitos de oxidação dos ácidos graxos, acidemias orgânicas e doenças de armazenamento do glicogênio, entre outras. |
| Urina | |
| Cetonas | A cetonúria, um aumento da excreção urinária de cetonas, pode ser encontrada de forma fisiológica na infância e na adolescência, quando não se acompanha de marcadores de estresse metabólico, como a acidose metabólica, a hiperlactatemia ou a hipoglicemia. Contudo, cetonúrias graves, com acidose associada, levantam a suspeita de um EIM. Já em recém-nascidos, a cetonúria não deve ser considerada um achado normal. |
| Tabela 2. Investigação bioquímica avançada para EIM | ||
| Método | Exemplos de EIM com alteração no teste | |
| Pesquisa de aminoácidos no plasma | Cromatografia líquida acoplada à espectrometria de massas em tandem | Doença da urina do xarope de bordo, fenilcetonúria, tirosinemia tipo 1, algumas doenças do ciclo da ureia, acidemias orgânicas |
| Pesquisa de ácidos orgânicos na urina | Cromatografia gasosa, tendo, como detector, o espectrômetro de massa (GC/MS) | Aminoacidopatias, acidemias orgânicas, distúrbios de oxidação dos ácidos graxos |
| Perfil de acilcarnitinas | Análise por injeção de fluxo, acoplada à espectrometria de massas em tandem (FIA-MS/MS) | Acidemias orgânicas e distúrbios de oxidação dos ácidos graxos |
Triagem neonatal diagnostica precocemente para tratar de forma mais efetiva
A triagem neonatal, também conhecida como teste do pezinho, tem a finalidade de detectar, em recém-nascidos pré-sintomáticos, anormalidades laboratoriais que permitem diagnosticar diversas doenças, incluindo muitos EIM.
Nos últimos anos, com os avanços nas técnicas laboratoriais, como o advento da espectrometria de massas em tandem, que possibilita a identificação de vários compostos em uma única análise realizada em poucos minutos, o número de doenças possíveis de rastrear foi significativamente ampliado. Ademais, com a utilização de técnicas de análise do DNA, alguns resultados bioquímicos anormais podem ser confirmados pela pesquisa molecular associada.
No Fleury, o teste de triagem neonatal ampliada é útil no diagnóstico de mais de 30 doenças tratáveis ou que podem ter sua evolução modificada com a detecção precoce. Está indicado para todos os recém-nascidos, independentemente de seu estado de saúde, devendo ser realizado a partir do segundo dia de nascimento e, preferencialmente, antes do quinto dia de vida.
Os recursos empregados conferem elevada sensibilidade e especificidade ao método, com redução do número de resultados falsamente anormais. Contudo, uma triagem neonatal normal não exclui completamente a presença de uma doença metabólica e, diante da suspeita clínica, a investigação laboratorial deve se impor.
Em cerca de 0,5% dos casos, pode haver necessidade da coleta de nova amostra para a complementação do estudo ou para a confirmação de resultados.
| Doenças pesquisadas pela triagem neonatal ampliada no Fleury: Aminoacidopatias: - Hiperfenilalaninemias - Leucinose - Hipermetioninemia - Homocistinúria - Hiperornitinemia - Citrulinemia- Tirosinemia Acidemias orgânicas: - Acidemias metilmalônicas e defeitos de síntese de cobalamina - Acidemias propiônicas - Acidemia glutárica tipo I - Deficiência de múltiplas desidrogenases (MADD ou acidemia glutárica II) - Acidemia 2-hidroxibutírica - Acidemia isovalérica - Acidemia arginossuccínica - Deficiência de metilcrotonil-CoA carboxilase - Acidemia metilglutacônica Defeitos da betaoxidação de ácidos graxos: - Deficiência de desidrogenase de acil-CoA de cadeia média (MCAD) - Deficiência de desidrogenase de acil-CoA de cadeia curta (SCAD) - Deficiência de desidrogenase de acil-CoA de cadeia longa (VLCAD) - Deficiência da 3-hidroxiacil-CoA desidrogenase de cadeia média (MCHAD/SCHAD) - Deficiência da 3-hidroxiacil-CoA desidrogenase de cadeia longa (LCHAD) - Deficiência de proteína trifuncional (TPD) - Deficiência de 3-metilcrotonil-CoA carboxilase - Deficiência de carnitina palmitoil transferase tipo 1 (CPT1) - Deficiência de 3-hidroxi-3-metilglutaril-CoA liase Galactosemias: - Deficiência de galactose-1-fosfato uridil transferase e variantes - Deficiência de galactoquinase - Deficiência de galactoepimerase Doenças hematológicas: - Hemoglobinopatias S (anemia falciforme), C e E - Deficiência de glicose-6-fosfato desidrogenase (G6PD) Doenças endocrinológicas: - Hipotiroidismo - Deficiência de 21-hidroxilase (hiperplasia adrenal congênita) Outras doenças: - Deficiência de biotinidase - Fibrose cística ou mucoviscidose - Imunodeficiência combinada grave (SCID) |
O papel das técnicas genético-moleculares
Os avanços em genética nos últimos anos tiveram grande impacto na compreensão dos EIM e na classificação dessas doenças. Devido à vasta heterogeneidade clínica e à complexidade da abordagem tradicional para a investigação desses quadros, a tecnologia do sequenciamento de nova geração (NGS) mudou o paradigma no diagnóstico de tais condições, que se beneficiaram com o advento dessa metodologia.
Para muitos EIM, não há um marcador específico ou, quando existe, pode estar associado a uma investigação invasiva, cara ou de pouca disponibilidade na rotina. Nesse contexto, os testes genéticos têm se mostrado frequentemente de grande utilidade para aumentar a acurácia diagnóstica, auxiliar a determinação prognóstica e permitir aconselhamento genético da família do indivíduo acometido. Ademais, na era da medicina personalizada, tal método pode contribuir para o advento de terapias genótipo-específicas.
No Fleury, o painel genético para EIM contempla a análise dos genes associados às principais doenças desse grupo, visando ao diagnóstico e à instituição de tratamento precoces. Pode ser indicado como recurso inicial para a pesquisa genética em pacientes com essas suspeitas.
Quando o painel genético específico não encontra uma variante patogênica que permita a conclusão diagnóstica, o sequenciamento do exoma, ou seja, de todos os éxons do genoma humano, constitui ainda um recurso importante na avaliação diagnóstica dos EIM. Realizado também por NGS, o exoma inclui a análise de SNV, indel e CNV, além da técnica de Sanger, que, quando aplicável, tem o objetivo de confirmar a presença e avaliar a segregação das variantes identificadas, utilizando uma análise conjunta dos resultados das amostras do paciente e dos seus genitores, se disponíveis.
| Ficha técnica Painel genético para EIM Metodologia: NGS, que permite a identificação de variantes de nucleotídeo único (SNV) e pequenas inserções e deleções (indel), bem como variações no número de cópias (CNV) que compreendam dois ou mais éxons dos genes estudados. Não inclui análise por MLPA. Amostra: sangue ou saliva Genes avaliados:ABCC8; ABCD1; ACADM; ACADVL; ACAT1; AGL; ALDH7A1; ALDOB; ARG1; ARSA; ARSB; ASL; ASS1; ATP7A; ATP7B; BCKDHA; BCKDHB; BCKDK; BTD; CBS; CPS1; CPT1A; CPT2; CTNS; CYP11B1; CYP21A2; CYP27A1; DBT; DLD; ETFA; ETFB; ETFDH; ETHE1; FAH; FBP1; FOLR1; G6PC; G6PD; GAA; GALE; GALK1; GALT; GAMT; GATM; GBA; GBE1; GCDH; GCK; GLA; GLB1; GUSB; GYS2; HADH; HADHA; HADHB; HLCS; HMGCL; HMGCS2; HPD; IDS; IDUA; INSR; IVD; KCNJ11; LIPA; LMBRD1; MMAA; MMAB; MMACHC; MMADHC; MOCS1; MPI; MTHFR; MTR; MTRR; MUT; NAGLU; NAGS; NPC1; NPC2; OTC; OXCT1; PAH; PCBD1; PCCA; PCCB; PGM1; PHGDH; PHKA2; PSAT1; PSPH; PTS; PYGL; QDPR; SGSH; SI; SLC16A1; SLC19A2; SLC19A3; SLC22A5; SLC25A13; SLC25A15; SLC25A20; SLC2A1; SLC2A2; SLC37A4; SLC46A1; SLC52A2; SLC52A3; SLC7A9; SMPD1; TAT; TCN2; TPP1. Prazo de resultados: em até 30 dias |
Como o NGS por ser incorporado ao algoritmo diagnóstico dos EIM
Os testes bioquímicos continuam tendo grande importância, uma vez que também ajudam a interpretar o painel molecular. Por outro lado, o NGS pode evitar exames invasivos em situações específicas. De modo geral, a pesquisa genética promove uma melhor acurácia ao diagnóstico:
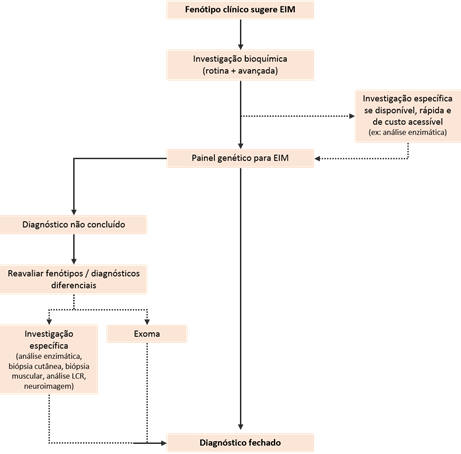
Adaptado de Ghosh A et al. ArchDisChild. 2017; 1-11.
Consultoria médica
Bioquímica Clínica
Dr. Gustavo Loureiro
Dr Nairo Sumita
Genética
Dr. Caio Robledo Costa Quaio
Dr. Wagner Antônio da Rosa Baratela
Neuroimagem
Dr. Carlos Jorge da Silva
Pediatria
Dra. Daniela Gerent Petry Piotto
Dra. Fernanda Picchi Garcia
Outros artigos
Leucemia mieloide aguda familiar
Exoma identifica nova variante patogênica germinativa no gene CEBPA, associado à doença
Estudos recém-publicados mostram relação causal entre vacinação contra herpes-zóster e redução de diagnósticos de demência
Estudos recém-publicados mostram relação causal entre vacinação contra herpes-zóster.
Ultrassonografia favorece o diagnóstico de condições que afetam a unha e suas estruturas
Ultrassonografia favorece o diagnóstico de condições que afetam a unha e suas estruturas.
Marcadores genéticos para doença hepática esteatótica podem contribuir para um manejo personalizado em casos selecionados
Marcadores genéticos para doença hepática esteatótica podem contribuir para um manejo personalizado.