A importância do exoma na investigação de síndromes genéticas
A crescente disponibilidade dos testes genéticos traz um grande avanço no diagnóstico desses quadros
Caso
O paciente do sexo masculino, 3 anos e 3 meses, foi encaminhado ao geneticista para investigação de quadro caracterizado por atraso global do desenvolvimento, baixa estatura proporcionada e baixo peso. Como antecedentes pessoais, tinha história de internação por bronquiolite aos 10 meses de vida, e infecções de vias aéreas de repetição durante o primeiro ano. Nasceu com 35 semanas, com baixo peso para a idade gestacional (peso = 1.610 g e comprimento = 40 cm), e apresentou cianose central, diagnosticada como síndrome do pulmão úmido, tendo permanecido em unidade de terapia intensiva neonatal por 17 dias para ganho ponderal.
O menor era filho único, de pais não consanguíneos. A mãe engravidou aos 29 anos (G1P0A0) e fez pré-natal adequado, com sorologias negativas para as infecções do grupo Torch e dengue. Negava uso de drogas lícitas ou ilícitas na gravidez. No quinto mês, desenvolveu uma adenomegalia axilar e evoluiu com insuficiência placentária, que resultou em restrição de crescimento intrauterino no feto. Não havia casos de síndromes conhecidas nem de deficiência intelectual na família.
Na consulta com o geneticista, a criança apresentou estatura de 71 cm (figura 1), peso de 7.040 g (figura 2) e perímetro cefálico de 42 cm (figura 3), tendo ficado determinado um déficit pôndero-estatural e microcefalia. Os dismorfismos faciais chamavam a atenção, especialmente a face triangular, a rarefação dos cabelos, o filtro nasal longo, o nariz, orelhas e olhos proeminentes, o hipotelorismo ocular e os dentes pequenos. Também foram observadas sobreposição bilateral de artelhos (2-3-4), clinodactilia bilateral dos quintos quirodáctilos e mancha hipercrômica na mão direita. A genitália era masculina com correção de hipospádia, sem sinais de desenvolvimento puberal – classificação de Tanner G1P1.
Figura 1
Gráfico de estatura para a idade em meninos de 0 a 5 anos (percentil) - Organização Mundial de Saúde
 Length/height (cm): comprimento / estatura (cm); age (completed months and years): idade (em anos e meses completos).
Length/height (cm): comprimento / estatura (cm); age (completed months and years): idade (em anos e meses completos).
Para melhor visualização da imagem, clique aqui.
Figura 2
Gráfico de peso para a idade em meninos de 0 a 5 anos (percentil) – Organização Mundial da Saúde
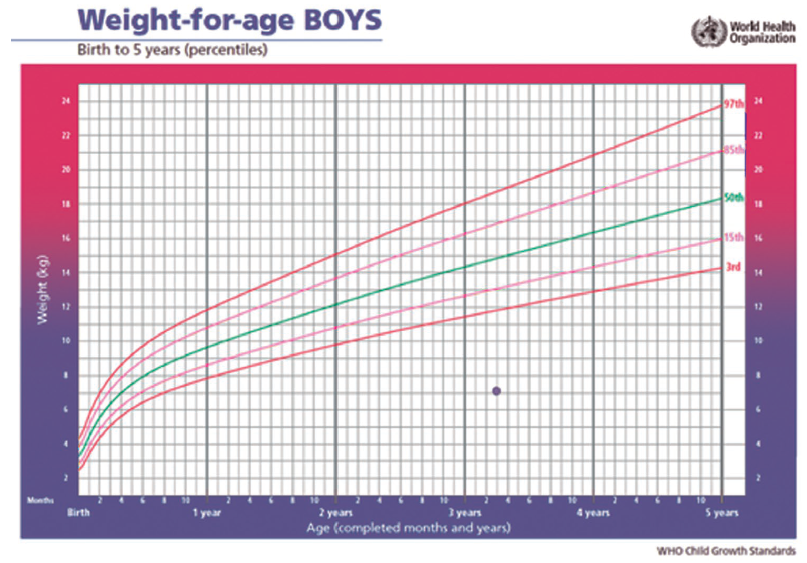 Weight (kg) = peso em kg; age (completed months and years) = idade (em anos e meses completos).
Weight (kg) = peso em kg; age (completed months and years) = idade (em anos e meses completos).
Para melhor visualização da imagem, clique aqui.
Figura 3
Gráfico de perímetro cefálico para a idade e meninos de 0 a 5 anos (percentil) – Organização Mundial da Saúde

Head circumference (cm) = perímetro cefálico em cm; age (completed months and years) = idade (em anos e meses completos).
Para melhor visualização da imagem, clique aqui.
Discussão
Na investigação de uma síndrome genética, a obtenção de uma história clínica minuciosa, incluindo antecedentes pessoais e familiares, e um exame físico detalhado configuram as principais ferramentas diagnósticas. Para afastar causas não genéticas, como trombofilias, endocrinopatias, doenças infecciosas ou infecções congênitas e doença vascular materna, entre outras, os exames laboratoriais e de imagem são recursos importantes. Em casos como o do paciente em estudo, alterações endocrinológicas associadas à baixa estatura, como a deficiência do hormônio de crescimento, igualmente devem ser pesquisadas. O paciente, entretanto, já havia feito um amplo estudo laboratorial prévio, incluindo ressonância magnética de crânio e hipófise, ultrassonografia de trato urinário, avaliação audiológica e diversos exames de análises clínicas, incluindo sorologias e dosagens hormonais sem alterações, além de cariótipo 46, XY e CGH-array sem achados patogênicos.
A contribuição genética
Em muitos casos referidos como quadros sindrômicos clinicamente reconhecíveis, a combinação das características fenotípicas deriva de uma alteração genética específica, permitindo um diagnóstico clínico. Contudo, a despeito de sinais e sintomas evidentes, em muitas circunstâncias não há como determinar a etiologia. Nessas situações, os testes genéticos se fazem relevantes.
Conjunto de todos os éxons do genoma humano, o exoma é a parte do genoma que contém as regiões codificadoras dos mais de 20.000 genes do corpo humano, na qual se encontra a maioria das alterações responsáveis pelas doenças genéticas. O sequenciamento dessa porção analisa detalhadamente essas regiões codificantes, de forma simultânea e a partir de uma única amostra de sangue ou saliva, constituindo um recurso importante na avaliação diagnóstica de muitas doenças de origem genética.
O teste tem especial indicação em pacientes cujo fenótipo não exibe o padrão de uma síndrome clínica bem definida ou em quadros com manifestações atípicas ou sintomas que se sobrepõem entre variadas condições. O caso apresentado se enquadrava nessas duas situações. O exoma também configurou uma alternativa relevante para a confirmação diagnóstica no paciente em avaliação porque outros testes feitos se mostraram negativos.
O estudo do exoma do paciente do caso avaliado identificou duas variantes, uma patogênica e outra provavelmente patogênica, em heterozigose, no gene LIG4, associado à síndrome LIG4 [OMIM:606593], de herança autossômica recessiva. A presença das variantes na criança foi confirmada por metodologia complementar Sanger e identificada, em heterozigose, nas amostras de ambos os genitores, o que indica que se encontravam em trans, ou seja, uma foi herdada do pai e a outra, da mãe (tabela 1).
Tabela 1. Variantes encontradas no exoma

Para melhor visualização da tabela, clique aqui.
O que é a síndrome LlG4?
A síndrome LIG4 é uma condição autossômica recessiva rara, causada por mutações no gene LIG4, localizado no cromossomo 13q22-q24, que codifica a DNA ligase IV – uma ligase nuclear distinta, que constitui um componente crítico do mecanismo de junção de extremidade não homóloga, necessário para o rearranjo V(D)J de segmentos gênicos durante o desenvolvimento normal de linfócitos e essencial para o reparo de quebras de fita dupla de DNA potencialmente letais em todos os tipos de células.
Grawunder et al. mostraram, em publicação de 1998, que a interrupção direcionada de ambos os alelos LIG4 em uma linhagem de células pré-B humana as tornou sensíveis à radiação ionizante e incapazes de completar o rearranjo V(D)J. Em camundongos, a homozigose para alelos nulos de LIG4 resultou em apoptose neuronal massiva e letalidade embrionária.
Os relatos mostram que pacientes com deficiência de ligase IV apresentam, como principais manifestações, microcefalia, características faciais incomuns, retardo de crescimento, atraso global de desenvolvimento e, tipicamente, pancitopenia. Ademais, observa-se radiossensibilidade pronunciada, em que as linhagens celulares exibem respostas de ponto de verificação do ciclo celular normais, mas comprometimento da quebra da fita dupla de DNA que se junta novamente.
Algumas questões ainda permanecem pendentes sobre a síndrome LIG4, especialmente se há predisposição a malignidade e imunodeficiências, como ocorre em outras doenças de instabilidade cromossômica. Em alguns casos relatados na literatura, foi observada leucemia linfoblástica de células T (Riballo et al., 1999), assim como mielodisplasia em um paciente (O'Driscoll et al., 2001). Tais achados indicam que a condição cursa com risco aumentado para neoplasias linfoides, possivelmente devido a aberrações cromossômicas resultantes de tentativas fracassadas de rearranjo. Vale, portanto, ressaltar que é imprescindível que os pacientes que evoluem para doenças malignas e apresentam características clínicas sugestivas de síndrome LIG4 tenham o diagnóstico considerado antes do início do tratamento, uma vez que são hipersensíveis aos efeitos da terapia.
Os principais diagnósticos diferenciais da síndrome LIG4 incluem a síndrome de quebras cromossômicas de Nijmegen (NBS), a síndrome de Seckel e a anemia de Fanconi (tabela 2).
Tabela 2. Comparação das características clínicas da síndrome LIG4 com os principais
diagnósticos diferenciais

Para melhor visualização da tabela, clique aqui.
Conclusão
Apesar da sobreposição fenotípica do caso descrito com a síndrome de Seckel, a presença de infecções de repetição na história clínica e a evolução durante o seguimento com pancitopenia, sugerindo radiossensibilidade celular pronunciada, foram indicadores úteis para apoiar o diagnóstico de síndrome LIG4, consoante aos achados do exoma.
A crescente disponibilidade dos testes genéticos traz um grande avanço no diagnóstico e na melhor caracterização das síndromes genéticas. A suspeita e a correta identificação de uma doença e o atual conhecimento de relações entre genótipo e fenótipo permitem prever as possíveis alterações clínicas presentes, proporcionando uma investigação consistente, intervenção precoce, aconselhamento genético e tratamento multidisciplinar oportuno das diferentes comorbidades, com impacto no prognóstico das doenças raras.
Dra. Mariana Carvalho Moreira
Médica Geneticista pelo HCFMUSP, Médica geneticista do Hospital Nove de Julho, Médica Geneticista doInstituto Jô Clemente de São Paulo (antiga Apae SP) e Médica Geneticista do Hospital São Camilo Unidades Santana/ Pompéia e Ipiranga
Consultoria Médica
Dra. Caroline Olivatti
[email protected]
Dr. Wagner Antonio da Rosa Baratela
Referências
Referências
1. Ben-Omran TI, Cerosaletti K, Concannon P, Weitzman S, Nezarati MM. A patient with mutations in DNA ligase
IV: clinical features and overlap with Nijmegen breakage syndrome. Am J Med Genet. 137A: 283-287, 2005.
2. Girard P-M, Kysela B, Harer CJ, Doherty AJ, Jeggo PA. Analysis of DNA ligase IV mutations found in LIG4
syndrome patients: the impact of two linked polymorphisms. Hum Molec Genet. 13: 2369-2376, 2004.
3. O'Driscoll M, Cerosaletti KM, Girard P-M, Dai Y, Stumm M, Kysela B, Hirsch B, Gennery A, Palmer SE, Seidel
J, Gatti RA, Varon R, Oettinger MA, Neitzel H, Jeggo PA, Concannon P. DNA ligase IV mutations identified in
patients exhibiting developmental delay and immunodeficiency. Molec Cell 8: 1175-1185, 2001.
Outros artigos
Ginecologista, o relógio biológico de sua paciente está correndo. Seu atendimento acompanha esse tempo?
Ginecologista, o relógio biológico de sua paciente está correndo.
Investigação de hiperprolactinemia e macroprolactina
Saiba como contornar os desafios na interpretação dos resultados da dosagem de prolactina
Strain atrial: uma nova ferramenta clínica para avaliação da função cardíaca
Strain atrial: uma nova ferramenta clínica para avaliação da função cardíaca
Nirsevimabe ajuda a prevenir a infecção e complicações causadas pelo VSR em lactentes
Nirsevimabe ajuda a prevenir a infecção e complicações causadas pelo VSR em lactentes