Contribuição da genética no diagnóstico de feocromocitoma/paraganglioma
Novo painel contempla grupos moleculares de variantes alélicas patogênicas ligadas a casos familiais
O feocromocitoma/paraganglioma (FEO/PGL) é uma neoplasia neuroendócrina rara, com incidência de 0,8/100.000 pacientes/ano. Trata-se de uma doença subestimada, com 50% dos casos diagnosticados em autópsias, a ponto de se estimar uma prevalência de 250-1300 casos/milhão em estudos de autópsias. A ocorrência é mais frequente entre a quarta e a quinta década de vida, tendo natureza benigna em 80-90% dos casos.
A doença se caracteriza pelo aumento na produção de catecolaminas pelas células cromafins, que, na maioria dos pacientes, tem origem na medula adrenal. Nos demais, a neoplasia está localizada fora da adrenal, como nos gânglios simpáticos do sistema nervoso autônomo, quando é designada paraganglioma (PGL) ou feocromocitoma extra-adrenal.
A apresentação clínica desse tumor caracteriza-se por um amplo espectro de sinais e sintomas, destacando-se a presença de paroxismos (tríade de cefaleia, palpitação e sudorese), acompanhada de hipertensão arterial (HAS) em 20-30% dos casos. É importante citar que o FEO/PGL responde por cerca de 0,1-1% dos casos de hipertensão secundária e por 0,1% dos novos casos de HAS que surgem anualmente.
Uma vez que o FEO/PGL pode ser esporádico ou hereditário, algumas informações da história clínica, do exame físico e do diagnóstico laboratorial são importantes para essa diferenciação.
Diferenças na evolução clínica e nas condutas dos feocromocitomas hereditários

Diagnóstico
A presença de níveis elevados de catecolaminas ou dos seus metabólitos (metanefrinas), resultantes da degradação tumoral, constitui a base do diagnóstico de FEO/PGL funcionantes. Atualmente, as dosagens das metanefrinas urinárias (24 horas) e plasmáticas devem ser os exames iniciais. No plasma, convém medir esses metabólitos preferencialmente por HPLC acoplada à espectrometria de massa em tandem, o padrão-ouro para o diagnóstico laboratorial, superior a qualquer combinação de exames para diagnóstico bioquímico de FEO/PGL. O encontro de metanefrinas plasmáticas (MLP) abaixo do valor de referência praticamente exclui FEO/PGL funcionantes, enquanto, na presença de dosagens 2-4 vezes acima do valor de referência, deve-se prosseguir a investigação com a cromogranina A ou o teste da clonidina com dosagens de metanefrinas plasmáticas.
Sugestão de fluxograma para investigação laboratorial

Clique na imagem para ampliá-la
O diagnóstico topográfico exige a realização da tomografia computadorizada (TC) de abdome com protocolo de adrenal e pelve, com ou sem contraste, devendo ser o exame inicial na investigação dessa neoplasia. Encontram-se valores de atenuação na fase pré-contraste superiores a 10 unidades Hounsfield (HU) e washout inferior a 60%. A ressonância magnética (RM) de adrenais e pelve constitui outro método diagnóstico topográfico com elevada sensibilidade diagnóstica (90-100%), evidenciando, na grande maioria dos casos, o hipersinal na sequência T2 e ausência de queda na intensidade do sinal na sequência out-of-phase. O método está indicado para os casos de FEO/PGL metastáticos e PGL de região cervical e intracardíaco, assim como na investigação de crianças, gestantes no segundo trimestre (sem contraste) e nos casos familiais.
Os métodos de Medicina Nuclear permitem melhorar o diagnóstico topográfico, funcional e de especificidade, como a cintilografia com MIBG marcada com iodo radioativo (131I ou 123I) e os exames de PET/CT com 68Ga-DOTA e com 18F-FDG, indicados quando há dúvida diagnóstica e nos casos familiais e metastáticos. O MIBG também tem aplicação teranóstica para pacientes com metástases que podem se beneficiar de tratamento com esse marcador. O estudo de PET-CT com 68Ga-DOTA ainda serve para casos metastáticos com indicação de protocolos de tratamento com lutécio (177Lu-DOTA).
Estudo genético para FEO/PGL
Cerca de 30-40% dos pacientes com FEO/PGL apresentam variante alélica patogênica germinativa e 46%, variante alélica patogênica somática. Evidentemente, a identificação de tais indivíduos depende de teste genético.
O Fleury dispõe de um painel genético para FEO/PGL e os candidatos para sua realização são:
- Jovens (<45 anos de idade)
- Antecedentes pessoais: hemangioblastoma de sistema de nervoso central, angiomas de retina, carcinoma renal de células claras, tumor neuroendócrino de pâncreas, neurofibromatose tipo 1, carcinoma medular de tiroide, hiperparatiroidismo primário, habitus marfanoide, neuromas mucosos, ganglioneuromatose intestinal, tumores pituitários e tumor estromal gastrointestinal
- História familiar de feocromocitoma/carcinoma medular de tiroide/síndrome de von Hippel-Lindau
- Lesão adrenal bilateral com valores de atenuação na fase pré-contraste portal >10 HU e washout <60% na TC
- Paraganglioma: chance de uma variante alélica patogênica em torno de 50-80%, mesmo sem histórico familiar
Três quartos de todos os casos de FEO/PGL de etiologia genética podem ter, como causa do quadro, um dos clusters/grupos moleculares a seguir:
- Cluster/grupo I: variantes alélicas patogênicas que levam ao fenótipo bioquímico noradrenérgico, produzindo noradrenalina e/ou dopamina e, consequentemente, seus metabólitos normetanefrina e 3-metoxitiramina, mas não adrenalina ou seu metabólito metanefrina.
- Cluster/grupo II: variantes alélicas patogênicas que geram o fenótipo bioquímico adrenérgico/metanefrina.
- Cluster/grupo III: variantes alélicas patogênicas desse grupo que causam tumores que expressam cromogranina A.
Características FEO/PGL de acordo com a mutação, apresentação clínica e evolução
Genes | Cluster 1 (relacionado ao ciclo de Krebs pseudo-hipóxico): SDHx (SDHA, B, C, D, F2), FH, MDH2 (10-15% de FEO/PGL) | Cluster 1 (relacionado ao VHL/EPAS1- pseudo-hipóxico): VHL, EPAS1/2 (HIF2A), PHD1/2 (15-20% de FEO/PGL) | Cluster 2 (relacionado à sinalização quinase): RET, NF1, MAX, TMEM127, HRAS (50-60% de FEO/PGL) | Cluster 3 (relacionado à sinalização Wnt): CSDE1, MAML3 (5-10% de FEO/PGL) |
| Percentual de mutação germinativa | 100% | 25% | 20% | 0% |
| Vias de sinalização | Ciclo de Krebs, pseudo-hipóxica, estabilização e sinalização HIF-2-alfa | VHL/EPAS1-pseudo-hipóxico | Quinase, PI3K/AKT, RAS/RAF/ERK, mTORC1/p70S6K | Wnt |
| Bioquímica | Normetanefrina, 3-metoxitramina | Normetanefrina | Normetanefrina e metanefrina ou apenas metanefrina | Normetanefrina, metanefrina |
| Imagem | 68Ga-DOTA-SSA PET-CT (exceto para FH) | 18F-DOPA PET-CT (possivelmente também para FH) | 18F-DOPA PET-CT | Desconhecida |
| Localização do tumor | Extra-adrenal, na maioria | Adrenal, extra-adrenal | Adrenal | Desconhecida |
| Risco metastático | Elevado a intermediário | Baixo a intermediário | Baixo | Intermediário |
| Idade de apresentação | Precoce (abaixo de 20-30 anos) | Precoce, alguns até na infância | Tardia, mas pode aparecer precocemente | Desconhecida |
Fonte: Nölting et al, 2019.
Principais genes identificados
Variantes alélicas patogênicas germinativas identificadas pelos casos de FEO/PGL familiais:
- VHL (von Hippel-Lindau), associado à síndrome de von Hippel-Lindau
- RET (rearranged during transfection) causando a síndrome de neoplasias endócrinas múltiplas tipo 2 (MEN 2)
- NF-1 (neurofibromatose tipo 1): associado à doença de von Recklinghausen (neurofibromatose tipo 1)
- Genes da síndrome dos paragangliomas familiares
Genes relacionados à síndrome dos paragangliomas familiares
| Localização | Fenótipo | Hereditariedade | Gene |
| 1p36.13 | Paragangliomas 4 | Autossômica dominante | SDHB |
| 1q23.3 | Paragangliomas 3 | Autossômica dominante | SDHC |
| 5p15.33 | Paragangliomas 5 | Autossômica dominante | SDHA |
| 11q12.2 | Paragangliomas 2 | Autossômica dominante | SDHAF2 |
| 11q23.1 | Paragangliomas 1, com ou sem surdez | Autossômica dominante | SDHD |
| 14q24.3 | Paragangliomas 7 | Autossômica dominante | DLST |
| 17p13.2 | Paragangliomas 6 | Autossômica dominante | SLC25A11 |
Variante alélicas patogênicas germinativas associadas com feocromocitoma/paraganglioma
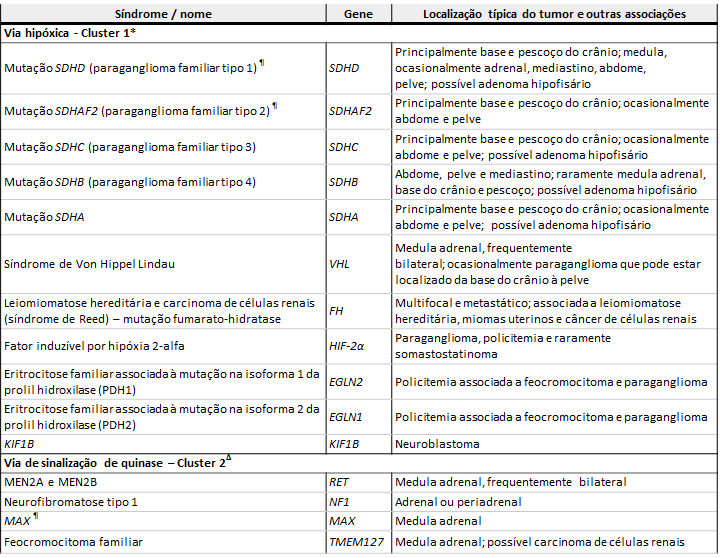
Clique na imagem para ampliá-la
Legendas:
GIST: tumor estromal gastrointestinal; MEN2: neoplasia endócrina múltipla tipo 2; SDH: succinato desidrogenase.
*Os tumores do cluster 1 são principalmente paragangliomas extra-adrenais (exceto na BVS, em que a maioria dos tumores está localizada na adrenal) e quase todos têm um fenótipo bioquímico noradrenérgico.
¶ Associada à impressão materna.
Δ Os tumores do cluster 2 são geralmente feocromocitomas adrenais com um fenótipo bioquímico adrenérgico.
Figura original modificada para esta publicação. WF jovem. Hipertensão endócrina. In: Williams Textbook of Endocrinology, Melmed S, Polonsky KS, Larsen PR, Kronenberg HM (Eds), 13ª ed, Elsevier Inc, Filadélfia 2015. p.556.
Referência
Nölting S; Ullrich M; Pietzsch J; Ziegler CG; Eisenhofer G; Grossman A; Pacak K. Current Management of Pheochromocytoma/Paraganglioma: A Guide for the Practicing Clinician in the Era of Precision Medicine. Cancers 2019, 11: 1505.
Consultoria médica
Endocrinologia
Dr. José G. H. Vieira
Dr. José Viana Lima Junior
Dra. Maria Izabel Chiamolera
Dra. Milena G. Teles Bezerra
Dr. Pedro Saddi
Dra. Rosa Paula Mello Biscolla
Dr. Rui M. B. Maciel
Genética
Dr. Caio Robledo Costa Quaio
Dr. Carlos Eugênio F. Andrade
Dra. Caroline Olivati
Dra. Daniele Paixão Pereira
Dr. Gustavo Marquezani Spolador
Dr. Wagner Antonio da Rosa Baratela
Medicina Nuclear
Dr. Marco A. C. Oliveira
Dra. Paola Emanuela P. Smanio
Imagem do Abdome
Dr. Ruy Rodrigues Galves Júnior
Outros artigos
Ginecologista, o relógio biológico de sua paciente está correndo. Seu atendimento acompanha esse tempo?
Ginecologista, o relógio biológico de sua paciente está correndo.
Investigação de hiperprolactinemia e macroprolactina
Saiba como contornar os desafios na interpretação dos resultados da dosagem de prolactina
Strain atrial: uma nova ferramenta clínica para avaliação da função cardíaca
Strain atrial: uma nova ferramenta clínica para avaliação da função cardíaca
Nirsevimabe ajuda a prevenir a infecção e complicações causadas pelo VSR em lactentes
Nirsevimabe ajuda a prevenir a infecção e complicações causadas pelo VSR em lactentes