Crescimento e baixa estatura
A baixa estatura requer avaliação detalhada do endocrinologista pediátrico e acompanhamento regular.
CASO CLÍNICO
Paciente do sexo feminino, 7 anos e 6 meses, passou em consulta com endocrinologista pediátrico, pois família referia que a filha era uma das menores da sala de aula, com crescimento lento. O interrogatório sobre diferentes aparelhos não trouxe outras queixas significativas.
A criança foi uma recém-nascida a termo, de uma gestação sem intercorrências, com peso de 2.590 g e comprimento de 49 cm. Recebeu aleitamento materno exclusivo até o primeiro mês de vida, quando iniciou aleitamento misto por baixo ganho de peso. Não teve alterações no desenvolvimento neuropsicomotor nem antecedentes de internações ou cirurgias. A vacinação estava em dia. Apresentava bom desempenho escolar, praticava atividade física três vezes por semana e mantinha alimentação adequada e variada.
Os pais eram saudáveis, assim como o irmão mais velho. A mãe tinha 162 cm de estatura e relatava menarca aos 11 anos e 6 meses. O pai tinha 180 cm e relatava desenvolvimento puberal dentro da normalidade. Os dados possibilitaram calcular a estatura-alvo da criança em 164,5 centímetros.
O exame físico inicial da criança não evidenciou alterações, com peso de 17,5 kg (Z-score = -2,41), estatura de 114 cm (Z-score = -2,01), pressão arterial (PA) de 90 x 60 mmHg e estadiamento puberal de Tanner M1P1.
Discussão
A baixa estatura (BE), uma das causas mais comuns de encaminhamento de pacientes ao endocrinologista pediátrico, é uma condição reconhecida desde a antiguidade, com citações de indivíduos com acondroplasia em sarcófagos e deuses egípcios baixos desde 360 a.C.
O crescimento consiste em um processo complexo, no qual nutrição, hormônios, fatores genéticos e ambientais têm importantes papéis, apenas com a menor parte das crianças apresentando alguma doença de base. O processo normal pode ser definido como um fenômeno Gaussiano, em que os valores da estatura recebem pontuação em uma curva de distribuição normal.
Nesse contexto, é fundamental a medida precisa da estatura e do peso com técnicas-padrão. Nos menores de 2 anos, o comprimento deve ser mensurado com a criança deitada e o resultado, colocado na curva da Organização Mundial de Saúde. Já em crianças acima dessa idade, a altura deve ser avaliada em pé e colocada na curva do Center for Disease Control, o CDC. O cálculo da velocidade de crescimento (VC) ao longo do tempo também auxilia a identificação de potenciais desvios da normalidade (quadro 1).
As alterações do crescimento devem ser expressas em desvios-padrão (DP) da média da população normal para crianças de mesmo sexo e idade. Em geral, crianças abaixo de 2 DP da média são classificadas como de BE. Esse valor usualmente corresponde ao percentil 3 da maioria dos gráficos de crescimento. Recomenda-se ainda a avaliação das proporções corporais por meio da relação segmento superior/segmento inferior, que pode ter utilidade para descartar doenças ósseas, como raquitismo e displasia.
Quadro 1. Valores de VC normal para idade
| ||||||||||||||||
A BE pode ocorrer no retardo do crescimento constitucional, na BE familiar, na desnutrição, no curso de doenças crônicas como anemia, doença renal, doença inflamatória intestinal e doença celíaca, em alterações endócrinas como a deficiência do hormônio de crescimento, a síndrome de Cushing e o hipotiroidismo, no retardo de crescimento intrauterino e em doenças genéticas diversas, com destaque para a acondroplasia, a síndrome de Turner e a síndrome de Noonan.
Mais recentemente, os defeitos da placa de crescimento também ganharam destaque, a partir do conhecimento da sua fisiologia, e se tornaram um potencial alvo para o desenvolvimento de novos tratamentos.
Investigação laboratorial da baixa estatura
Uma avaliação completa dos pacientes com BE, com o principal objetivo de afastar doenças crônicas, é importante. A tabela abaixo destaca as principais investigações diagnósticas e os exames laboratoriais indicados na abordagem inicial.
| Tabela 1. Investigação inicial da criança com BE para afastar doenças crônicas | |
| Condição clínica | Exame |
| Descartar anemia | Hemograma |
| Avaliar desnutrição | Ferritina e albumina |
| Descartar doença hepática crônica | Enzimas hepáticas (TGO, TGP, gama-GT) |
| Pesquisar doenças inflamatórias crônicas | VHS |
| Descartar doenças osteometabólicas | Cálcio, fósforo, fosfatase alcalina |
| Descartar doença renal crônica e acidose tubular | Creatinina, ureia, sódio, potássio, gasometria venosa e urina tipo 1 |
| Investigar doença celíaca | Anticorpos antitransglutaminase e IgA sérica |
| Descartar alteração na função da tiroide | TSH e T4 livre |
| Avaliar o eixo GH/IGF-1 | IGF-1 e IGFBP-3 |
| Descartar síndrome de Cushing | Cortisol urinário de 24 horas e/ou cortisol salivar das 23 horas (ambos em duas amostras) e/ou teste de supressão de cortisol com dexametasona overnight |
| Excluir síndrome de Turner | Cariótipo com banda G em todas as meninas |
| Avaliar presença de verminose | Protoparasitológico de fezes |
O hormônio de crescimento
Sintetizado, armazenado e liberado pela hipófise anterior em resposta ao estímulo do hormônio liberador do GH (GHRH), o hormônio de crescimento (GH) chega aos tecidos-alvo através da corrente sanguínea e responde pela maioria dos processos metabólicos e de crescimento no organismo.
A ação do GH é mediada pelo IGF-1, um outro hormônio secretado pelo fígado, que estimula a divisão e o crescimento celular. A análise do eixo do GH/IGF-1 deve sempre fazer parte da avaliação clínica inicial, pois qualquer distúrbio que o afete pode ocasionar anormalidades no crescimento. Devido à secreção pulsátil do GH, sua concentração pode apresentar grande variação mesmo em indivíduos sem alterações do eixo GH/IGF-1, fazendo com que a dosagem do IGF-1 - que apresenta secreção mais estável - seja bem mais confiável nesse contexto.
Testes de estímulo para avaliação do GH
Para os pacientes que apresentam fatores sugestivos de deficiência de GH, com valor de IGF-1 reduzido para a idade ou mesmo dentro da normalidade, recomenda-se a realização de testes de estímulo para avaliação do pico de GH. Há diversas opções de exames funcionais para avaliar a reserva secretória de GH, cada qual com sua particularidade, relacionada a uma maior ou menor eficácia do estímulo, efeitos colaterais e contraindicações (veja tabela 2).
| Tabela 2. Testes de estímulo para avaliação do GH | |||||
| Exame | Racional | Como é feito | Interpretação | Efeitos adversos | Contraindicações |
| Teste de intolerância à insulina |
|
|
|
|
|
| Teste de estímulo de GH com clonidina |
|
|
|
|
|
| Teste de estímulo de GH com glucagon |
|
| Respostas normais: - GH acima de 3 microg/L em adultos com IMC até 25 kg/m2 - GH acima de 1 microg/L em adultos com IMC acima de 25 kg/m2 - GH acima de 1 microg/L para dose fixa de glucagon - GH acima de 2 microg/L para a dose por peso |
|
|
Avaliação por imagem
Raios X para determinação da idade óssea
A avaliação dos centros de ossificação da mão e do punho permite verificar a sequência de aparecimento e morfologia dos centros de ossificação das falanges, metacarpos, ossos do carpo, rádio e ulna, assim como a fusão das epífises com as diáfises, o que possibilita examinar a predição do crescimento e desenvolvimento do paciente. O método de avalição de Greulich & Pyle é o mais utilizado e consiste em um atlas com padrões de idade óssea desde o nascimento até os 19 anos, para os meninos, e até os 18 anos, para as meninas.
A comparação da idade óssea com a cronológica é fundamental no diagnóstico diferencial de baixa estatura. Crianças com variação da normalidade, como o retardo constitucional de crescimento e puberdade ou a aceleração constitucional de crescimento e puberdade, podem ter um atraso ou um avanço na idade óssea, respectivamente. Condições patológicas costumam apresentar idade óssea com mais de 2 DP para a média da idade.
Ressonância magnética da região hipofisária
Método não invasivo, a ressonância magnética (RM) apresenta grande sensibilidade na identificação de anormalidades anatômicas ou patológicas da região hipotálamo-hipofisária, como a presença de hipoplasia hipofisária no paciente com deficiência de GH. Tem sido considerada útil não apenas no diagnóstico, mas também na decisão terapêutica do paciente com deficiência de GH.
O protocolo FAST ou de cortes rápidos pode ser utilizado como triagem em casos de hipopituitarismo em substituição à RM de hipófise convencional. O exame tem curta duração e dispensa o uso de contraste endovenoso, sendo bem tolerado mesmo por crianças pré-adolescentes. O objetivo é avaliar alterações volumétricas da adeno-hipófise (como a hipoplasia), as alterações morfológicas da haste hipofisária (como a interrupção abrupta ou espessamentos anômalos) e a caracterização da topografia da neuro-hipófise, com vistas a demonstrar a integridade da comunicação física do eixo hipotálamo-hipofisário – em situações com interrupção da haste hipofisária, por exemplo, o hipersinal habitual da neuro-hipófise é ectópico, junto ao recesso infundibular do terceiro ventrículo.
DESFECHO
A análise dos antecedentes pessoais da criança em estudo mostrou que não havia restrição de crescimento intrauterino, já que seu peso ao nascimento fora maior que 2.500 g.
Nos primeiros meses de acompanhamento, mesmo com boa VC, a paciente se mantinha próxima da curva do Z-score -2 e com projeção da estatura final abaixo do padrão familiar e da estatura-alvo (figura 1), o que levou o endocrinologista a investigar possíveis causas que estivessem impedindo um crescimento dentro do esperado. Foram, então, realizados exames gerais (tabela 3), que não mostraram alterações, testes de estímulo da liberação do GH (tabelas 4 e 5) e exames de imagem (figura 2), assim como cariótipo, com resultado 46,XX (50 metáfases analisadas).
| Tabela 3. Exames bioquímicos, metabólicos e hormonais em diferentes faixas etárias da paciente | ||||
| | 7a 6m | 10a 2m | 10a 8m | Valor de referência |
| Hemoglobina (g/dL) | 14,3 | 12,0 – 14,5 | ||
| Hematócrito (%) | 43,1 | 36,0 – 43,0 | ||
| Leucócitos (células/m3) | 8.510 | 3.400 – 10.800 | ||
| Plaquetas (células/m3) | 460.000 | 150.000 – 450.000 | ||
| Glicose (mg/dL) | 90 | 98 | 96 | 70 – 99 |
| IGF-1 (ng/mL) | 119 | 395 | 317 | 7a 6m: 91 – 414 10a: 156 – 670 |
| IGFBP-3 (ng/mL) | 4.800 | 2.019 – 5.515 | ||
| FSH (UI/mL) | 7,2 | 4,7 | ||
| LH (UI/mL) | 0,6 | 1,7 | Pré-pubere: <0,3 | |
| T4 livre (ng/dL) | 1,5 | 1,4 | 1,4 | 1,0 – 1,7 |
| Estradiol (ng/dL) | <0,5 | 1,6 | 0,6 – 2,7 | |
| TSH (mUI/L) | 3,2 | 5,6 | 4,4 | 0,6 – 5,4 |
| PCR (mg/dL) | <0,03 | |||
| ATPO (UI/mL) | <34 | <34 | ||
| ATG (UI/mL) | < 115 | <115 | ||
| Anticorpos antitransglutaminase tecidual IgA, antigliadina IgA, antigliadina IgG, antiendomísio IgA e antiendomísio IgG | Não reagente | |||
| K (mEq/L) | 4,9 | 3,5 – 5,0 | ||
| Na (mEq/L) | 142 | 137 – 148 | ||
| Ferro (mcg/dL) | 87 | 50 – 120 | ||
| Ferritina (mcg/L) | 52 | 10 – 150 | ||
| Ureia (mg/dL) | 34 | 10 – 50 | ||
| Creatinina (mg/dL) | 0,44 | 0,5 – 1,0 | ||
| TGO (U/L) | 27 | Até 32 | ||
| TGP (U/L) | 15 | Até 33 | ||
| 25-hidroxivitamina D (ng/dL) | 27 | 30-60 | ||
| ATPO: anticorpo antiperoxidase tiroidiana; ATG: anticorpo antitiroglobulina. | ||||
| Tabela 4. Teste de tolerância à insulina (8 anos de idade) | ||||||
| GH (microg/L) | 0,29 | -- | 1,78 | 0,33 | 0,12 | 0,11 |
| Glicemia (mg/dL) | 88 | 20 | 15 | 51 | 92 | 82 |
| Tempo (min) | 0 | 15 | 30 | 60 | 90 | 120 |
| Tabela 5. Teste de GH após estímulo com clonidina (8 anos de idade) | ||||
| GH (microg/L) | 0,99 | 2,32 | 4,38 | 1,33 |
| Tempo (min) | 0 | 60 | 90 | 120 |
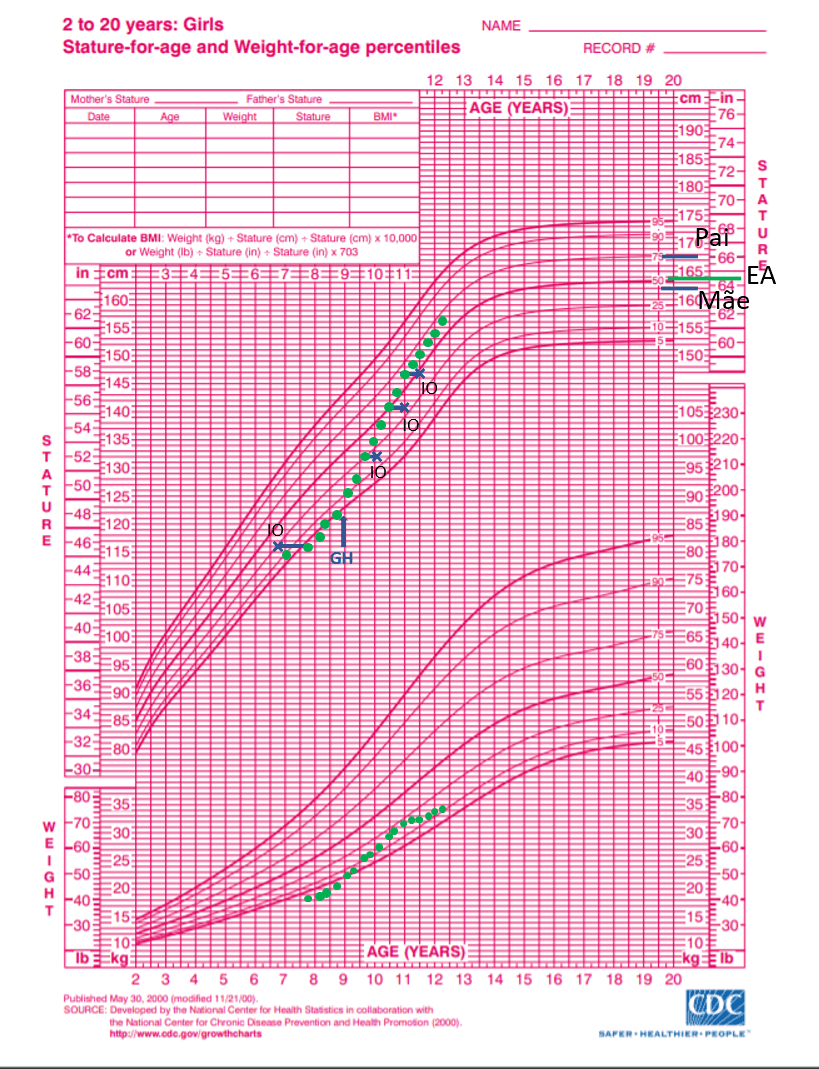
Figura 1. Gráfico de curva de estatura para idade, em meninas. Estatura-alvo (EA): 164,5 cm
CONCLUSÃO
A decisão de tratar uma criança com BE é complexa e deve considerar fatores fisiológicos e não fisiológicos, como o entendimento dos pais sobre os efeitos psicológicos da condição em seu filho. Alguns acreditam que a BE pode causar baixa autoestima, baixo desempenho escolar ou dificuldade nos relacionamentos. Entretanto, há pouca evidência de que o tratamento com GH possa melhorar o desempenho psicológico.
As diretrizes terapêuticas podem variar ao redor do mundo, mas o diagnóstico e o início do tratamento precoces são cruciais, principalmente para as crianças candidatas ao uso de GH.
No caso descrito, apesar da queixa inicial, da observação de baixa liberação de GH nos testes de estímulo e do gráfico de crescimento, a família optou por aguardar algum tempo antes de iniciar a reposição do GH, que teve início quando a criança estava com 8 anos e 10 meses.
Se, antes do GH, a VC da paciente era de 5,6 cm/ano, passou para 9 cm nos primeiros 11 meses de uso do hormônio na dose de 0,12 UI/kg/dia. De fato, o maior ganho de estatura ocorre geralmente no primeiro ano de tratamento.
A criança em estudo apresentou pubarca aos 9 anos e 4 meses e telarca aos 10 anos e 2 meses, comprovada por valor púbere de hormônio luteinizante. Até o momento da última avaliação, com 12 anos e 4 meses, não havia apresentado menarca.
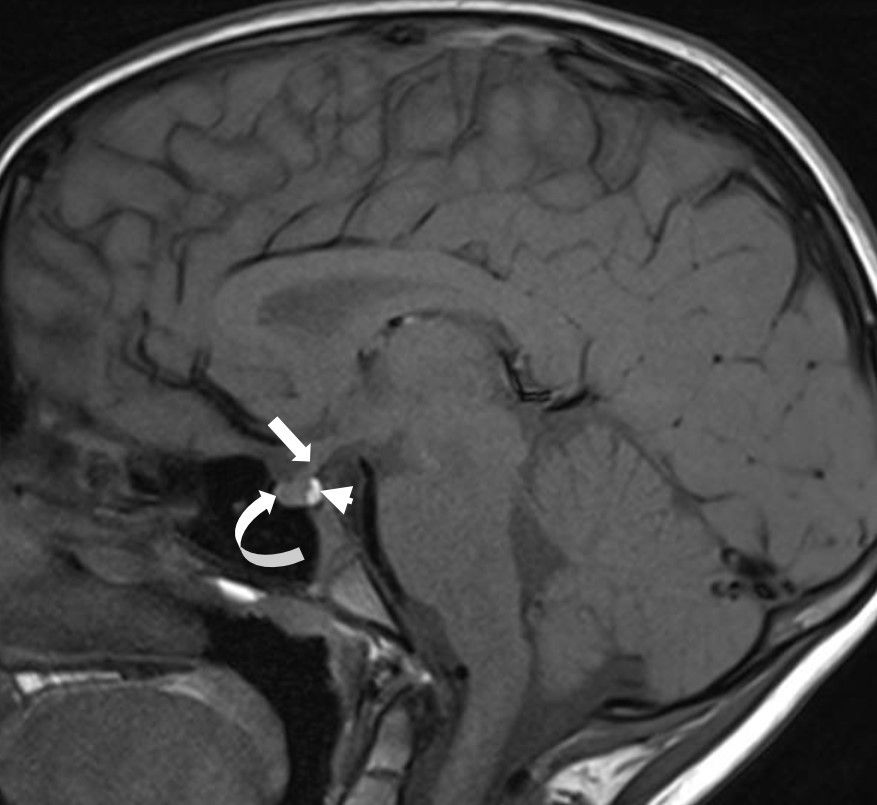
Figura 2. RM de sela turca, corte sagital, T1, protocolo FAST, realizada aos 8 anos, na qual se observam a haste hipofisáfia (seta), a adeno-hipófise (seta curva) e a neuro-hipófise (cabeça de seta), sem alterações.
Em que pensar diante da suspeita de baixa estatura de causa genética?
| Síndrome de Turner Síndrome genética associada à perda completa ou parcial de um dos cromossomos X, no sexo feminino. Está relacionada a baixa estatura, puberdade atrasada, disgenesia ovariana, hipogonadismo hipergonadotrófico, infertilidade, cardiopatia congênita e doenças autoimunes. |
| Síndrome de Noonan Condição genética caracterizada por alterações faciais, baixa estatura, atraso no desenvolvimento neuropsicomotor, malformação congênita (cardiopatia) e distúrbio de coagulação. Cerca de 50% dos casos apresentam mutação no gene PTPN11. Os genes BRAF, CBL, HRAS, KRAS, MAP2K1, MAP2K2, NF1, NRAS, RAF1, RIT1, SHOC2, SOS1, SPRED1 e LZTR1 também foram implicados na gênese da síndrome e podem ser avaliados por meio de um painel genético, disponível no Fleury. |
| Baixa estatura por defeito em fatores de transcrição – hipopituitarismo O desenvolvimento hipofisário depende de diversos fatores de transcrição, razão pela qual um defeito em qualquer um deles pode acarretar diferentes deficiências hormonais. O POU1F1 e o PROP1 são os principais fatores de transcrição que causam deficiência de GH, porém defeitos nos fatores HEX1, LHX3, LHX4, SOX2 e SOX3 igualmente podem estar envolvidos. |
| Baixa estatura por defeito em genes reguladores da placa de crescimento Para que os ossos cresçam, é necessário que haja um processo de ossificação endocondral na cartilagem de crescimento. Falhas em qualquer um dos principais genes reguladores dessa estrutura, como SHOX, NPR2, NPPC, ACAN, IHH e FGFR3, podem causar baixa estatura associada a uma alteração das proporções corporais. |
| Baixa estatura por defeitos em genes que regulam o eixo GH/IGF-1 Defeitos em qualquer um destes genes costumam estar associados a uma baixa estatura isolada: GH1, GHSR, GHR, STAT5B, IGF1, IGF1R, IGFALS e PAPP-A2. |
O material foi elaborado pelas endocrinologistas pediátricas do Fleury Kids:
Dra. Patrícia Débora Cavalcanti Tosta Hernandez
Dra. Vanessa Radonsky
Consultoria Médica
Endocrinologia
Dr. José Viana Lima Junior
Dra. Maria Izabel Chiamolera
Dra. Rosa Paula Mello Biscolla
Pediatria
Dra. Daniela Gerent P. Piotto
Dra. Fernanda Picchi Garcia
Radiologia – Idade Óssea
Dr. Shri Krishna Jayanthi
Radiologia Pediátrica
Dr. Rodrigo Regacini
Neuroimagem
Dr. Antonio Carlos Maia Junior
Dr. Carlos Jorge da Silva
Dr. Carlos Toyama
Dr. Lucas Avila Lessa Garcia