Perguntas e respostas sobre o teste pré-natal não invasivo
O NIPT é o teste identifica DNA fetal livre na circulação materna. Saiba mais sobre o exame
Assista ao vídeo e leia o conteúdo abaixo:
Por que o teste é chamado de NIPT?
A sigla vem do inglês, noninvasive prenatal testing, ou teste pré-natal não invasivo.
O teste pode ser solicitado como NIPT mesmo?
Sim. Outra possibilidade é pedir como identificação de aneuploidias fetais no sangue materno ou, ainda, com os nomes dos testes comerciais das respectivas empresas. No Fleury, o exame é da Ilumina e tem o nome, no exterior, de Verify.
Como é obtido o resultado do teste?
O teste identifica DNA fetal livre na circulação materna. A maioria desse DNA fetal livre vem da placenta e a menor parte, do feto. Dessa forma, fragmentos de DNA fetal livre são analisados por sequenciamento, com posterior quantificação das frações de DNA dos cromossomos examinados em relação a um padrão. Um algoritmo de bioinformática é usado para a liberação do resultado.
Que síndromes são avaliadas no NIPT?
São avaliadas a trissomia do cromossomo 21 (síndrome de Down), a trissomia do 18 (síndrome de Edwards), a trissomia do 13 (síndrome de Patau) e as alterações dos cromossomos sexuais: monossomia do X (síndrome de Turner), XXY (síndrome de Klinefelter), XXX e XYY.
Qual a sensibilidade para a detecção dessas síndromes?
As taxas de detecção mostram-se elevadas, chegando perto de 98% para as trissomias do 21, 18 e 13, com baixos índices de falso-positivos (veja tabela abaixo).
| Taxa de detecção | Taxa de falso-positivo | Valor preditivo positivo | |
| Trissomia do 21 (síndrome de Down) | 99,7% | 0,04% | 78% |
| Trissomia do 18 (síndrome de Edwards) | 97,9% | 0,04% | 62% |
| Trissomia do 13 (síndrome de Patau) | 99% | 0,04% | 35% |
| Monossomia do X | 95,8% | 0,14% | 31% |
| Não monossomia do X | 93% | 0,14% | >90% |
| T21 (gemelar) | 93,7% | 0,23% | 60% |
Adaptado de: Gil et al 2017.
É possível coletar o exame com 9 semanas de gestação?
A idade gestacional da coleta do NIPT deve ser sempre a partir de 10 semanas. Antes disso, a fração fetal é baixa e, portanto, há maior chance de falha do teste e maior risco de necessidade de recoleta. Como a fração fetal aumenta com a idade gestacional, independentemente da técnica utilizada pelo teste, antes de 10 semanas seu teor sempre será baixo. Por outro lado, não há limite superior de idade gestacional para a coleta do exame.
O que é a fração fetal (FF)?
A estimativa de FF é um componente do algoritmo do exame, que, em associação com outras métricas de qualidade, determina a confiança nos resultados, embora não seja usada isoladamente para excluir amostras. Recomenda-se um mínimo de 4% para uma boa acurácia do teste. Apesar de o ponto de corte de 4% não ser mais tão consensual como antes, ainda se preconiza que valores inferiores a 4% sejam repetidos, o que, no Fleury, é feito sem ônus.
Que fatores interferem na FF?
Os fatores incluem idade gestacional inferior a 10 semanas, obesidade, condições associadas à placenta pequena (triploidia, trissomia do cromossomo 18 e trissomia do cromossomo 13), problemas na coleta, no envio e no armazenamento do sangue, fertilização in vitro e múltiplos da mediana de PAPP-A e βHCG livre muito baixos.
Qual a conduta diante de um resultado do NIPT com FF abaixo de 4%?
Pela técnica da quantificação utilizada no Fleury, é possível liberar resultados com FF inferior a 4%. No entanto, a acurácia desse teste em relação à identificação das alterações pode estar diminuída. Um exemplo típico é a triploidia. Como todos os cromossomos examinados estão igualmente triplicados, o teste não consegue detectar a alteração e o resultado pode ser de ausência de aneuploidia com uma FF baixa (em geral inferior ou igual a 2%).
O que checar diante de FF baixa
- A idade gestacional da coleta deve ser de 10 semanas ou mais.
- A ultrassonografia precisa ser feita para confirmar se a gestação ainda é viável, principalmente se o exame anterior tiver sido realizado antes das 9 semanas.
- Quando a gestante for obesa, convém aguardar pelo menos duas semanas para tentar repetir o teste.
- Aguardar a ultrassonografia morfológica do primeiro trimestre para avaliar a presença de alterações fetais sugestivas de cromossomopatias, sobretudo triploidia, trissomia do 18 e trissomia do 13.
Qual a chance de obtenção do resultado na recoleta?
A chance fica em torno de 70%. Devido aos fatores que interferem na FF, uma parte importante dos resultados continua com fração baixa ou inconclusiva.
Qual a frequência de FF baixa?
No Fleury, a taxa de FF inferior a 4% é de aproximadamente 1,6%, abaixo da descrita na literatura – de 3% a 6%. A literatura reporta índices de recoleta maiores no sequenciamento pela técnica do SNP (cerca de 5%).
Que conduta deve ser tomada diante de um resultado positivo?
Depende da alteração encontrada, que deve ser interpretada com os dados da idade materna e os achados ultrassonográficos. De qualquer forma, é importante lembrar que o NIPT é um teste de rastreamento, não um teste diagnóstico. Sua chance de acerto, ou seja, o valor preditivo positivo (VPP), varia conforme a gestação, se de alto ou baixo risco.
Para a trissomia do cromossomo 21, dada a alta sensibilidade e o elevado VPP do teste, a confirmação diagnóstica pode ser oferecida por biópsia de vilo corial ou amniocentese, independentemente dos achados ultrassonográficos. Já para as trissomias dos cromossomos 13 e 18, que estão associadas a marcadores e malformações fetais importantes, deve-se considerar os dados da ultrassonografia.
Dessa maneira, na presença de alterações fetais no estudo de imagem, a confirmação diagnóstica pode ser realizada por biópsia de vilo corial. Nos casos de ultrassonografia morfológica do primeiro trimestre, incluindo aqueles com translucência nucal normal, o cariótipo fetal em líquido amniótico coletado por amniocentese é melhor. Nessa situação, a chance de o resultado alterado derivar de mosaicismo placentário, e não de alteração fetal, é maior, devendo-se, portanto, evitar a punção placentária da biópsia de vilo corial. O mesmo ocorre quando o NIPT é positivo para a síndrome de Turner (monossomia do X).
Se estiver diante de translucência nucal aumentada, observada em cerca de 80% dos casos dessa condição, a confirmação pode ser feita pela biópsia de vilo corial, pois o VPP do teste nessa situação é muito alto. Se a translucência nucal estiver normal, a chance de falso-positivo do teste aumenta, por mosaicismo placentário, dando-se preferência para o estudo do cariótipo no líquido amniótico.
Nas gestações com cromossomos sexuais alterados, como XXY, XXX e XYY, a opção do diagnóstico invasivo deve ser ponderada diante das baixas repercussões observadas na evolução clínica de indivíduos com essas alterações. No entanto, o VPP do teste aparentemente independe dos achados ultrassonográficos e a chance de acerto do NIPT é elevada.
Para que gestantes devo pedir o NIPT?
Solicitando o teste para todas as gestantes, considerando a síndrome de Down como base, aproximadamente 98% dos casos seriam detectados como positivos, mas o falso-positivo seria de 1%. Com base em fatores de risco, a taxa de detecção para síndrome de Down ficaria entre 75% e 85%, porém com falso-positivo menor (0,4%). O NIPT pode ser realizado em todas as gestantes, principalmente quando a população atendida tem idade materna avançada e o nível de ansiedade é elevado.
Fatores de risco que constituem indicação para o NIPT
- Idade materna maior ou igual a 35 anos
- Translucência nucal aumentada
- Gestação anterior acometida por aneuploidias avaliadas no NIPT
- Pais com translocação robertsoniana balanceada, com risco aumentado de trissomia do 21 ou do 13
- Rastreamento combinado com risco final de até 1 em 1.000
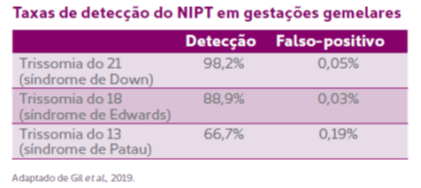
O NIPT pode ser feito na gestação gemelar?
Pode. Os dados apontam que a acurácia do teste se assemelha à encontrada nas gestações únicas. Contudo, a casuística dos estudos em gemelares ainda é limitada, notadamente quando comparada com as gravidezes únicas. Em gestações trigemelares ou mais, não há dados para embasar seu uso.
É possível realizar o exame pós-FIV e ovodoação?
Sim, o NIPT pode ser feito em gestações pós-FIV, seja com feto único, seja em gravidez gemelar, bem como nos casos de ovodoação. No entanto, se for gestação gemelar pós-ovodoação, a técnica de SNP não pode ser utilizada. O Fleury não emprega essa metodologia, mas o sequenciamento massivo por quantificação.
Em caso de gestação gemelar, o teste permite saber o feto afetado?
Não. Nenhum teste de NIPT disponível para uso diagnóstico é capaz de identificar qual o feto gemelar afetado por trissomia. Diante de um resultado positivo nessas situações, a ultrassonografia morfológica é fundamental para identificar marcadores sugestivos de trissomias e, com isso, indicar o feto suspeito.
É possível identificar o sexo dos fetos na gestação gemelar pelo NIPT?
A técnica utilizada no Fleury procura detectar a presença do marcador genético masculino – o cromossomo Y. Dessa forma, pode identificar se há um feto masculino ou não. Pela técnica de SNP, não adotada pelo Fleury, aparentemente é possível determinar o sexo de cada feto, mas com acurácia pouco conhecida.
Em que situações a biópsia de vilo corial ou a amniocentese para cariótipo fetal não devem ser substituídas pelo NIPT?
Em casos de malformações fetais, de translucência nucal igual ou superior a 3,5 mm, de suspeita de triploidia fetal e de infecções congênitas. Essas condições contraindicam o uso do NIPT pelo risco muito elevado de alteração genética, que já justifica a indicação direta do cariótipo fetal invasivo, inclusive, muitas vezes necessitando de extração de DNA para realização de teste molecular por arrays ou de exoma.
Por que o NIPT não detecta triploidia?
Na triploidia, todos os cromossomos estão triplicados. Com isso, pela técnica de quantificação do DNA fetal usada no Fleury, não haveria nenhum cromossomo com fração aumentada sobre o outro para ser identificado. Também na triploidia digênica, de origem materna, que é a que cursa com malformações fetais graves e, por isso, faz parte do diagnóstico pré-natal relacionado à Medicina Fetal, a placenta é extremamente pequena, com consequente FF muito baixa. Frações fetais reduzidas diminuem a confiabilidade do teste e são ainda responsáveis pelos resultados inconclusivos. Pela técnica de SNP, não utilizada no Fleury, a triploidia digênica também não é detectada. Essa metodologia até permite o diagnóstico da triploidia diândrica, de origem paterna, que está associada à gestação molar. No entanto, esses casos são passíveis de diagnóstico ultrassonográfico, dispensando o NIPT.
O que significa o resultado inconclusivo?
Significa que não foi possível estimar se o risco para aneuploidias é baixo ou alto na amostra analisada. Pode estar relacionado principalmente com FF baixa, decorrente dos fatores já mencionados. Existem casos de inconclusivos que são diferentes dos relacionados à FF. O sequenciamento aponta que pode haver alguma anormalidade no genoma indicado que não é detectada por esse teste, razão pela qual a Associação Americana de Genética recomenda ultrassonografia morfológica e aconselhamento genético para discussão de testes adicionais. Nessa situação, pode haver alguma síndrome genética, em geral relacionada a microdeleções ou a uma dissomia uniparental. Contudo, não há razão para repetição do NIPT e o teste genético adicional seria invasivo para realização de microarray no líquido amniótico.
Nos raros casos de resultado inconclusivo, a equipe médica do Fleury entra em contato com o médico para discutir o direcionamento diagnóstico mais adequado. A gestante também pode marcar consultoria em Medicina Fetal para receber explicações da equipe médica ou mesmo marcar aconselhamento genético com os geneticistas. Essas consultas devem ser agendadas pela Central de Atendimento do Fleury.
O que o NIPT ampliado avalia a mais?
Além das alterações numéricas nos cromossomos analisados pelo NIPT convencional, o teste avalia o risco de aneuploidias em todos os outros cromossomos e de algumas síndromes genéticas mais raras, associadas a microdeleções subcromossômicas.
Quais as indicações do NIPT ampliado?
Não existem recomendações de sociedades médicas para a realização do NIPT ampliado. Mas, em caso de solicitação, o teste poderia ser reservado para casos de restrição do crescimento fetal abaixo do percentil e de cardiopatia fetal cornotruncal, pela associação com síndrome de DiGeorge (deleção 22q11.2). Outra possível indicação seria nos casos de NIPT inconclusivo por alteração em alguma parte do genoma. Tradicionalmente, o próximo passo, nesses casos, seria solicitar o procedimento invasivo, mas, quando a gestante não aceita realizar a biópsia de vilo corial ou a amniocentese, o NIPT ampliado pode ser indicado na repetição do exame, pois investiga, além dos cromossomos 13, 18, 21, X e Y, todos os demais cromossomos e mais cinco síndromes genéticas raras. Nessa situação, já houve casos em que o teste ampliado revelou trissomia dos cromossomos 7 e 22.
Qual a performance do NIPT ampliado?
Para as trissomias dos cromossomos 13, 18 e 21 e alterações dos cromossomos sexuais, é semelhante ao NIPT. Para as síndromes gênicas avaliadas, a taxa de detecção varia de 60% a 70%, embora a chance de o teste confirmar a alteração seja baixa (ou seja, o VPP é de aproximadamente 5%). Ainda há necessidade de estudos adicionais e maior casuística para definir as taxas de sensibilidade, especificidade, falso-positivos e falso-negativos do teste.
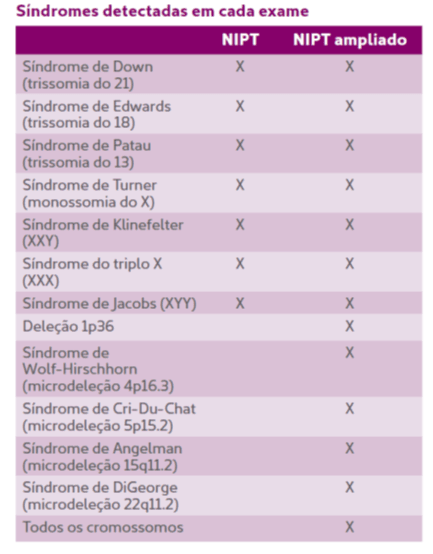
POR DENTRO DAS ALTERAÇÕES AVALIADAS PELO NIPT/NIPT AMPLIADO
Síndrome de Down
Trata-se da aneuploidia compatível com a vida mais comum, causada pela presença de uma cópia extra do cromossomo 21. Sua incidência é de 1:700 nascimentos e se eleva com o avanço da idade materna. Por ser a síndrome mais incidente, configura o principal motivo pelo qual as gestantes procuram os serviços de diagnóstico pré-natal.
A condição associa-se com deficiência intelectual, cardiopatias e alterações digestivas, entre outras manifestações.
Síndrome de Edwards
Ocasionada pela presença de cópia extra do cromossomo 18, está associada a altas taxas de abortamento e malformações graves, que levam a uma elevada mortalidade neonatal. Tem incidência de 1:5.000 nascimentos.
Síndrome de Patau
Decorre da presença de cópia extra do cromossomo 13 e correlaciona-se com taxas elevadas de abortamento e malformações graves. As principais malformações encontradas são cerebrais (holoprosencefalia alobar) e faciais (fenda labial e palatina mediana, probóscide, ciclopia) e cardiopatias, entre outras. A sobrevida além do primeiro ano de vida é extremamente rara. Está presente em 1:16.000 nascimentos.
Síndrome de Turner
Com incidência variando de 1:5.000 a 1:8.000 nascidos vivos do sexo feminino, caracteriza-se por um cromossomo X e deleção total ou parcial do segundo X, associando-se a abortamentos. Após o nascimento, a presença de estigmas “turnerianos”
(estrabismo, hipertelorismo ocular e mamário, implantação baixa de orelhas e cabelos, micrognatia, pescoço curto e/ou alado, etc.) ajuda o clínico a levantar a suspeita. As meninas acometidas têm retardo no crescimento desde a vida intrauterina. Assim, de 95% a 100% das pacientes exibem baixa estatura.
Síndrome de Klinefelter
A condição afeta apenas os homens e é marcada pela presença de uma ou mais cópias extras do cromossomo X. Constitui-se na anormalidade cromossômica mais comum, com incidência de 1:500 a 1:800 nascimentos do sexo masculino.
Muitos pacientes são assintomáticos. Nos que possuem sinais clínicos, observa-se esterilidade, desenvolvimento mamário, dificuldades sociais e de aprendizado, alta estatura e sobrepeso, entre outros.
Síndrome do XXX
Considera-se a anomalia cromossômica mais comum nas mulheres, acometendo 1:1.000 nascimentos do sexo feminino. As manifestações incluem baixa estatura, epicanto, hipotonia e clinodactilia. Em idade escolar, as crianças podem apresentar maior chance
de déficits cognitivos e dificuldades de aprendizado. Existe a possibilidade de a trissomia não ser diagnosticada, visto que algumas pacientes têm a doença em grau leve ou assintomática.
Síndrome do XYY
Aneuploidia dos cromossomos sexuais causada pela presença de cópia extra do cromossomo Y, incide em 1:1.000 nascimentos do sexo masculino. No período neonatal, as manifestações são mais sutis, incluindo clinodactilia, hérnia inguinal e pectus carinatum. No começo da infância, as crianças afetadas apresentam alta estatura, atraso na fala, déficit de aprendizado e alterações de comportamento. Na maioria dos pacientes, a síndrome não afeta a fertilidade e não ocasiona alterações significativas, motivo pelo qual permanece sem diagnóstico.
Síndrome da deleção 1p36
Também conhecida como monossomia 1p36, é considerada umas das síndromes de deleção cromossômica mais comuns. Caracteriza-se por alterações craniofaciais, retardo no desenvolvimento, déficit intelectual, alterações de comportamento, hipotonia, problemas de alimentação e epilepsia. Sua incidência varia de 1:5.000 a 1:10.000 nascidos vivos.
Síndrome de Wolf-Hirschhorn
Com incidência de 1:50.000 nascimentos, acomete duas vezes mais o sexo feminino que o masculino, sendo ocasionada pela microdeleção 4p16.3. Os portadores exibem microcefalia, hipertelorismo, glabela proeminente, nariz largo, filtro labial curto, micrognatia, orelhas displásicas e lábio leporino. Também são observados retardo mental de graus variados, baixo peso ao nascimento, hipotonia, convulsões, surdez, baixa estatura e malformações cardíacas, renais e genitais.
Síndrome de Cri-Du-Chat
Decorre da microdeleção 5p15.2 e está presente em 1:50.000 nascimentos. Além do choro característico ao nascer, que se assemelha ao choro de gato em sofrimento, outras manifestações incluem microcefalia, baixo peso ao nascimento, hipertelorismo e assimetria facial, bem como retardo mental e do crescimento.
Síndrome de Angelman
Tem incidência de 1:10.000 a 1:40.000 nascimentos e deriva da microdeleção 15q11.2. Uma característica da condição é a facilidade, do portador, de sorrir e rir, observada muito cedo. Muitos lactentes não mostram sinais da doença ao nascimento e o retardo do desenvolvimento aparece entre 6 meses e 1 ano de idade, em conjunto com hipotonia, hiperatividade, risadas explosivas e prejuízo da linguagem verbal. O retardo mental é grave e 80% das crianças apresentam convulsões antes de 4 anos de idade. Observam-se ainda marcha atáxica e tremores em extremidades. Os pacientes acometidos atingem um nível de desenvolvimento de apenas o equivalente a 24 a 30 meses de idade.
Síndrome de DiGeorge
Causada pela microdeleção 22q11.2, ocorre em 1:3.000 nascidos vivos. Em neonatos, as manifestações englobam dismorfismos craniofaciais, malformações de vias aéreas, cardiopatias e malformações renais. A síndrome causa alterações neurológicas e comportamentais em lactentes e crianças maiores, assim como atraso no desenvolvimento da linguagem e disfunções do sistema imunológico.
Consultoria médica
Medicina Fetal
Dra. Ellen Beatriz Araujo Freire
[email protected]
Dr. Fernando Takashi Kojima Marques
[email protected]
Dr. Javier Miguelez
[email protected]
Dra. Luciana Carla Longo e Pereira
[email protected]
Dr. Mário H. Burlacchini de Carvalho
[email protected]
Genética
Dr. Caio Robledo C. Quaio
[email protected]
Dr. Carlos Eugênio Fernandez de Andrade
[email protected]
Dr. Wagner Antonio da Rosa Baratela
[email protected]
Outras publicações
18/6/2026
O que é o teste Oncotype DX?17/6/2026
Síndrome de Li-Fraumeni